Menu
close
PAH is a rare disease that is defined by a progressive increase in pulmonary vascular resistances due to the development of constrictive vascular lesions within the patients’ lungs. This remodeling is mostly characterized by proliferation of smooth muscle cells within the arterial medial layer, proliferation and recruitment of fibroblasts and myofibroblasts within the adventitial, medial and intimal/neointimal layer, and to some extent endothelial proliferation and angiogenesis. A moderate but constant chronic inflammatory perivascular infiltrate is present in such lesions, sometimes evolving to vasculitis-like infiltrates. The role of inflammation in the natural history of the disease has been subject to much debate in the past and has recently led to novel experimental therapeutic approaches and one phase 2 trial (Toshner M et al. Eur Respir J. 2022; 59(3):2002463).
In our previous work, we have histomorphologically confronted lungs of patients with idiopathic pulmonary arterial hypertension (IPAH) with samples from patients suffering from autoimmune connective tissue disease-associated PAH (CTD-PAH), both conditions that are classified into group 1 of the diagnostic Nice-classification. This and other subsequent studies have shown that within the entity of PAH, different compartments of the pulmonary vasculature are involved, including severe and inflammatory remodeling of the post-capillary compartment, resembling to pulmonary veno-occlusive disease (PVOD), a rare subentity which occurs more frequently in patients with CTD-PAH (Dorfmüller P et al. Hum Pathol. 2007; 38(6):893-902). These observations have led to the last update of the Nice-classification where PVOD has been moved to group 1 and is now defined as PAH with overt features of venous and capillary involvement (Simonneau G, t al. Eur Respir J. 2019; 53(1):1801913).
Other than the local expression of T-cell-recruiting chemokines, such as Fractalkine (CX3CL1) in endothelial cells of pulmonary arterial lesions in PAH patients, we have found increase in number and reactivity of its receptor CX3CR1 on circulating CD4+CD8+ T-Lymphocytes of these patients, underscoring a possible role for pro-inflammatory mechanisms in PAH (Dorfmüller P et al. Am J Respir Crit Care Med. 2002; 165(4):534-9) (Balabanian K et al. Am J Respir Crit Care Med. 2002; 165(10):1419-25). In addition, smooth muscle cells of PAH patients display increased amounts of CX3CR1, and its ligand CX3CL1 induces proliferation of PAH SMCs in vitro, indicating a direct impact of pro-inflammatory mediators on the vascular remodeling process (Perros F et al. Eur Respir J. 2007; 29(5):937-43). Of interest, systematic immunophenotyping of human diseased lungs suggests that the inflammatory process is highly organized with perivascular tertiary lymphoid tissues/tertiary follicles, perivascular dendritic cells and local immunoglobulin class switching, observations that appear to argue for specific immune-adaptive mechanisms (Perros F et al. Eur Respir J. 2007; 29(3):462-8) (Perros F, et al. Am J Respir Crit Care Med. 2012; 185(3):311-21).
With regard to the variable involvement of different parts of the lung vasculature in group 1 of the Nice-classification our group has provided evidence for the involvement of the systemic lung vasculature (bronchial vessels and vasa vasorum) in group 1 PAH and also in group 4 PH (chronic thromboembolic PH) (Ghigna MR et al. Eur Respir J. 2016; 48(6):1668-1681) (Dorfmüller P et al. Eur Respir J. 2014; 44(5):1275-88). It is now widely recognized that plexiform lesions and so called SiMFis lesions in PAH correspond to hyperplastic anastomoses between the pulmonary and the systemic vasculature. SiMFis lesions are more frequently encountered in PAH patients with BMPR2 mutations and the hypertrophy of bronchia vessels is also the cause for more frequent hemoptysis in this subgroup. Interestingly, venous remodeling can be observed in both, PAH and CTEPH (group 4), a secondary effect that is due to the connection of the low-pressure system of post-capillary vasculature to the high-pressure regime of systemic vessels within the lung that originate directly or indirectly from the aorta.
On a molecular level, we have shown that GCN2, the protein product of inheritable PVOD-gene EIF2AK4, is decreased or abolished in both, PVOD and PAH, as well as their respective experimental models, arguing one more time in favor of a continuum of pulmonary vascular diseases in PH, ranging from predominant arterial constriction to veno-occlusive remodeling (Nossent EJ et al. J Heart Lung Transplant. 2018; 37(5):647-655).
New directions of interest of this tissue-oriented pathobiology lab are coming from our previous findings and will focus on the spatial distribution of lung and heart involvement in PAH group 1, including IPAH and PVOD, as well as group 4 (CTEPH), but also much more prevalent group 2 PH (PH associated with left heart disease). Since the remodeling of muscular-type arteries in PH, especially the occurrence of plexiform lesions in PAH, appears to be a secondary phenomenon, and because microvascular changes in smallest arterioles, venules and the capillary bed are a common denominator in many subgroups of PH, we will address these lung vascular compartments, combining visual and molecular in situ-tools, associating TEN X transcriptome spatial analysis and protein ligation assays (protein-protein-interaction assessment) with AiryScan-equipped confocal microscopy and automated micro-morphometry. Three-dimensional understanding of an initial remodeling process within the smallest vascular structures of pulmonary and systemic lung vessels appear paramount in pulmonary vascular medicine, where vasodilating substances like endothelin-receptor antagonists or soluble guanylate cyclase stimulators are in clinical use, despite the evidence of irreversible fibrotic lesions of medium-sized arteries in PAH and obliterative thromboembolic lesions in CTEPH. With regard to human lung samples, we are collaborating with our colleagues from the French Referral Center for Severe Pulmonary Hypertension at Bicêtre University Hospital, Paris Saclay University (Prof. Marc Humbert), in addition to the DZL Biobank in Giessen. Our rodent experimental models include Sugen/hypoxia (PH/PAH), Monocrotaline (PH), pulmonary venous banding (LHD-PH, PVOD), and the recently established pulmonary arterial ligature model (CTEPH).
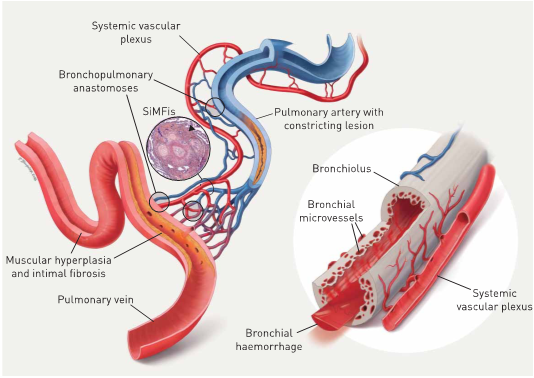
Impact of hypertrophic systemic vasculature in pulmonary arterial hypertension (PAH): an explanatory approach. The pulmonary artery (top centre, blue) and the adjacent bronchiole (right, grey) are covered by a hypertrophic systemic vascular plexus, comprising systemic arterial (red) and venous (blue) vessels and microvessels. The systemic plexus entertains several anastomoses with the pulmonary artery, the capillary bed and the pulmonary vein (bottom left, red): these bronchopulmonary anastomoses appear to bypass a constricting PAH-lesion, represented by medial thickening and intimal fibrosis (centre). In our view, the observed singular millimetric fibrovascular lesions (SiMFis) (photo inset) corresponds to a transversal section through a dense association of a (remodelled) pulmonary artery and several hypertrophic systemic vessels. Eventually, the increased systemic blood flow into arterioles, capillaries and the pulmonary vein leads to structural changes of the latter: muscular hyperplasia and focal intimal fibrosis within the pulmonary vein are observed. The hypertrophic systemic vasculature has a parallel impact on the adjacent airway (right): an increase in size and number of bronchial vessels and subepithelial microvessels leads to intrabronchial haemorrhage and eventually haemoptysis (from: Ghigna MR et al. Eur Respir J. 2016; 48(6):1668-1681).
Soni Savai-Pullamsetti: TBX4 mutations in human PAH; distinct global transcriptional regulatory landscape in small vessel vasculopathy of CTEPH.
Herbert Schiller: Spatially resolved analysis of sub-tissular niche proteomes and associated cell circuit states in the distal human lung.
PD Dr. Dr. Peter Dorfmüller
Universitätsklinikum Gießen/Marburg
Instistut für Pathologie
Langhansstr. 10
35392 Giessen
Tel: +49 (0) 641 985 41102 (Sek.)
Tel: +49 (0) 641 985 41116 (Office)
Fax: +49 (0) 641 985 41119
Email:
Medical students: Anna Rotert, Marvin Strauch, Patrick Bohne